Two-Photon Microscopy
Introduction to two-photon excitation microscopy
Two-photon excitation microscopy is a particularly microscopy technique based on the capability, under specific circumstances, to excite with two photons one electron in the ground state. The particularity of this phenomenon concern the fact that the light used to induce the excitation has a lower wavelength than the collected fluorescent. This makes two-photon microscopy particularly cell-friendly and allows among others, to image living specimens with little to no physiological alterations.
Two-photon microscopy of in vivo brain function
Legend : Two-photon microscopy of in vivo brain function.
- (a) Basic mechanism of two-photon fluorescence.
- (b) Schematic of surgical preparation of exposed cortex, with sealed glass window and microscope objective positioning. Green dot shows location of two-photon fluorescence.
- (c) Examples of two-photon maps of the vasculature following intravenous injection of dextran-conjugated fluorescein. Black dots and stripes show red blood cell motion. (
- d) Dual-channel imaging of neuronal (green) and vascular (red) signals: (left) Oregon Green 488 BAPTA-1 AM calcium sensitive dye stained neurons and (right) transgenic mouse expressing green fluorescent protein (GFP) in a subpopulation of neurons (mouse supplied by Jeffrey M. Friedman, Rockefeller University, New York) [101]. Texas dextran red is the intravascular tracer in both cases.
- (e) Three channel imaging of Tg2576 APP Alzheimer’s disease mouse model with amyloid-targeting dye (blue), GFP expressing neurons and dendrites (green) and vasculature (red). Source https://commons.wikimedia.org/wiki/File:Two-photon_microscopy_of_in_vivo_brain_function.jpg adapted from [1] and contributed by Elizabeth Hillman (Columbia University, New York).
Principle of two-photon microscopy
Two-photon microscopy is based on the principle originally predicted by M. Goeppert-Mayer in 1931 and then demonstrated by W. Kaiser and C.G.B Garrett in 1961. The phenomenon concerns the absorption of two photons of identical or different wavelengths in order to excite a molecule. Most of fluorescence microscopy techniques are based on single photon absorption (Figure 2). In single photon absorption the electron of a fluorophore is excited by a wavelength that has more energy than the one emitted by the fluorescence phenomenon. Traditionally UV excitation is used to collect blue fluorescence, blue light to excite fluorophores that emit in the green and so on. In two photon microscopy the electron of the fluorophore is excited by two photons with a longer wavelength than the resulting fluorescence emission (Figure 2). Contrary to single absorption, two-photon absorption is more difficult to obtain at lower light intensity since it is a second-order process. On the other hand when photons are dense enough two-photon absorption prevails over the linear single absorption.
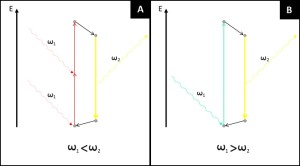
Figure 2. Difference between standard single photon absorption of fluorescence microscopy and two-photon absorption of two-photon microscopy. (A) In single photon absorption an electron hit by a photon with sufficient energy to excite it jumps to an higher energetic level. Through internal conversions the excited electron lose part of its energy (non-radiative transition, black arrow) before eventually emit fluorescence. The energy of the exciting photons is always bigger than the emitted one. (B) In two photon absorption an electron is moved from the ground level by a first photon, to reach the excited state the photon must absorb a second photon while the electron is in its virtual state. The energy of the two photons is completely absorbed by the electron (normally in its ground state) and the emitted fluorescence has a shorter wavelength than the exciting light.
Two-photon microscopes: main advantages
The optical scheme of a two photon microscope is not much different from a standard laser scanner confocal microscope. In both technologies a spotted light is produced by a laser source passing through a pinhole. Two galvanic mirrors then manipulate the angle of the light in order to scan the sample. The main differences concern the lack of the sample pinhole and the illumination source.
In confocal microscopy sample pinhole is in charge of filtering the fluorescence that originates at the focal plane. In two-photon microscopy this is not needed thanks to the different phenomenon used to excite fluorophores (Figure 3). The physical principle of two-photon absorption dictates that only at high photon density the phenomenon prevails over one photon absorption. The relation is a second order relation and implies that only when photons are concentrated enough fluorescence occurs (Figure 4). This brings to two photon microscopy a better resolution in the z axis when compared to confocal microscopes.
The second difference is related to the laser. While confocal microscopes laser emission is rather constant, two-photon microscopy uses pulsed laser in order to concentrate as much photons as possible in each single pulse.
The way the microscope illuminates the sample in a two-photon microscopy brings several advantages when compared to confocal microscopy. The main advantage is that two-photon microscopy can produce fluorescence with wavelengths (far-red, infrared) that are less subjected to light scattering. This significantly enhances the capability of the microscope to penetrate deeper in dense tissue. Longer wavelengths are also less toxic for cells and moreover the probability of secondary photochemical reactions are significantly much lower than what observed in confocal microscopy.
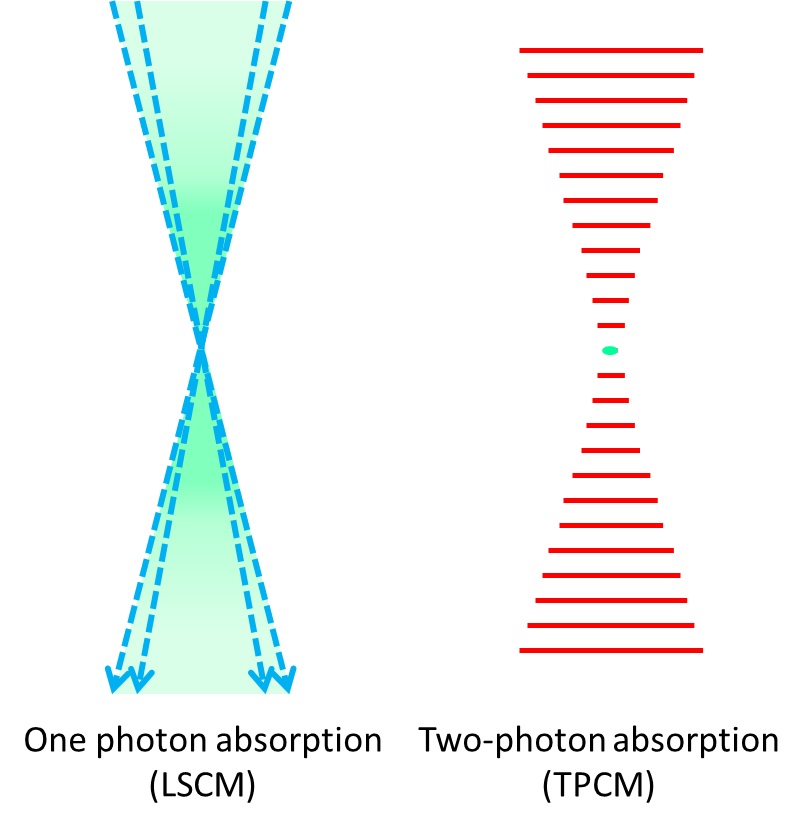
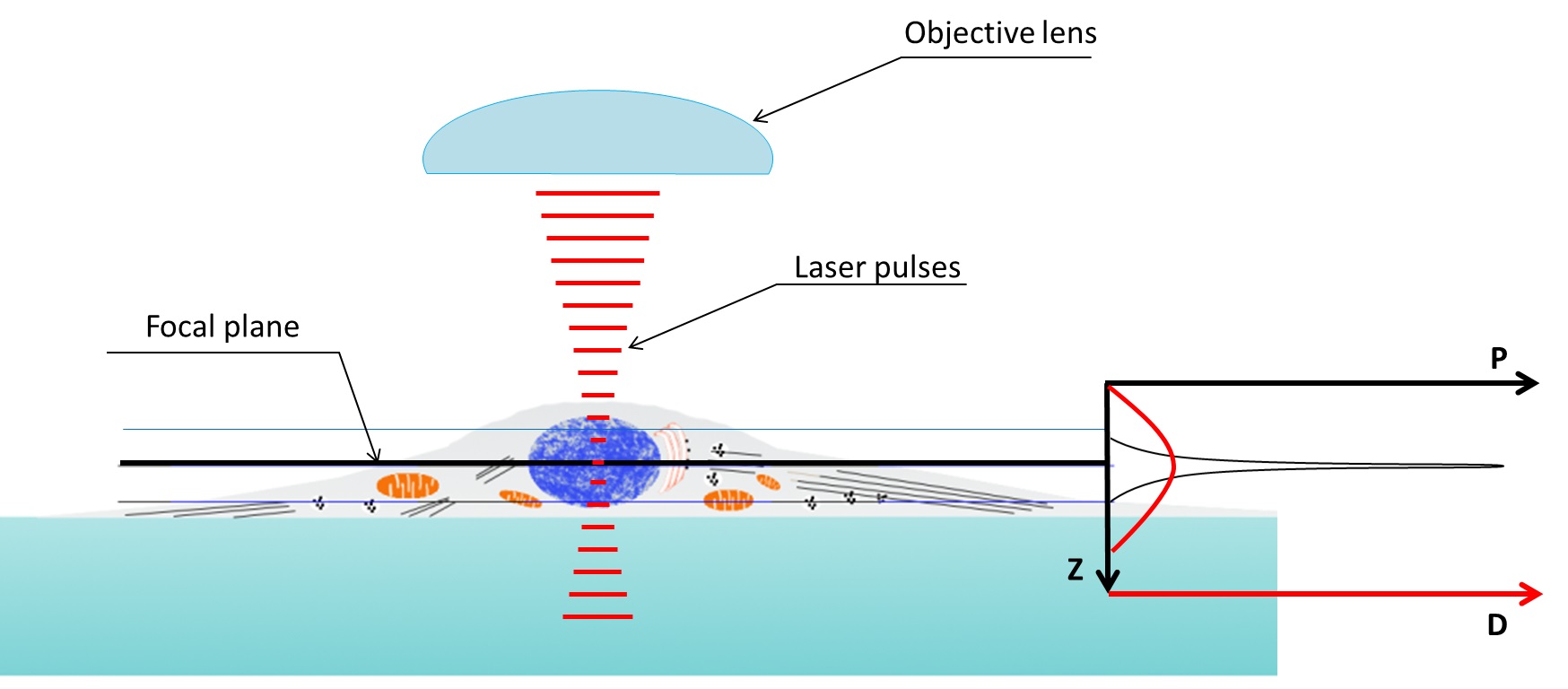
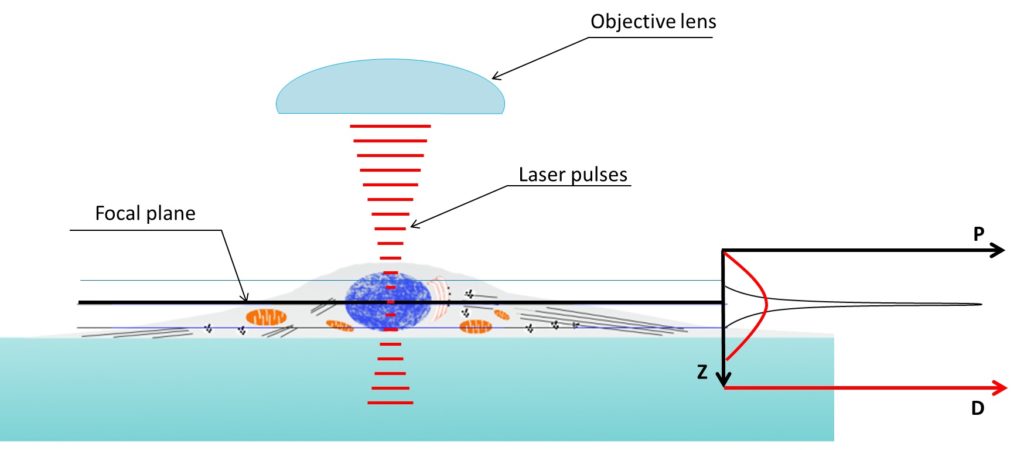
Figure 3. Excitation (in green) provided by a laser scanning confocal microscope (LSCM) blue laser (left) and two-photon microscope red-light pulses (right). Fluorescence produced in the z axis by confocal microscopy illumination is mostly concentrated at the focal plane, on the other hand two-photon microscopes are able to produce fluorescence in a very narrow spot.
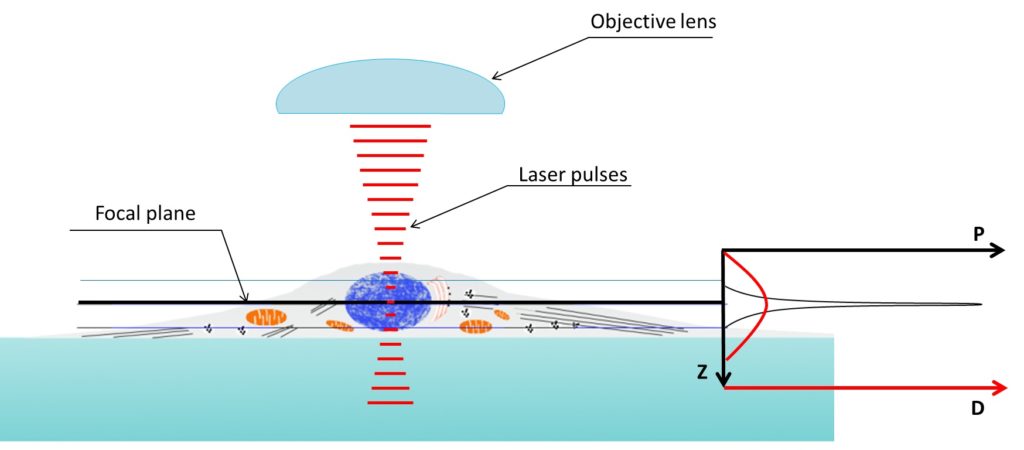
Figure 4. Two photon microscopy z axis resolution is much better than confocal microscopy. Density of the photons used to illuminate reach a peak in the focal plane. Two-photon absorption probability (P, graph on the right) only prevail above a specific threshold depending on the photon density (D, graph on the right). Being the relation between photon density and probability of two-photon absorption not linear the capability of exciting an electron (i.e. obtain fluorescence) is very limited in the z axis.
Reference
[1] Hillman EMC. Optical brain imaging in vivo: techniques and applications from animal to man. Journal of biomedical optics. 2007;12(5):051402. doi:10.1117/1.2789693.